PNC-27
For in vitro testing and laboratory use only. Not for human or animal consumption. Bodily introduction is illegal. Handle only by licensed professionals. Not a drug, food, or cosmetic. Educational use only.
PNC-27: Bold Anticancer Concept, Still a Research Story
PNC-27 is not a "secret cancer cure," but a synthetic chimeric peptide created from a p53-derived segment and a penetratin sequence, so the interest in it grew out of a quite serious anticancer logic rather than empty hype. In published preclinical studies, it was linked to HDM-2 in the membranes of tumor cells and described as a molecule that, in the research context, caused rapid membrane damage and cell lysis rather than classical apoptosis.
In cell-based, ex vivo, and some animal models, PNC-27 showed notable antitumor activity, and in certain studies there was also interest in combinations, for example with paclitaxel. But the whole honest intrigue is that this story still remains mainly preclinical: there is no convincing clinical basis in humans, which means that turning it into a promise of a "working treatment" would be far too bold.
That is exactly why PNC-27 is compelling not as a ready-made solution, but as an unusual and genuinely intriguing research platform in oncology. For a client, this is the kind of case where attention is drawn not by loud advertising, but by the idea itself — unconventional, bold, and still much more scientific than clinically confirmed.
PNC-27: A Scientific Review
Based on peer-reviewed literature — see References. Last updated: April 2026.
The Short Version
PNC-27 is a 32-amino acid chimeric anticancer peptide that kills cancer cells by a mechanism unlike any approved drug: it binds to HDM-2 (the human homologue of MDM2) expressed on the outer surface of cancer cell membranes, assembles with it into transmembrane pores, and causes the cancer cell to rupture from within in a form of cell death its discoverers have named “poptosis.” Normal cells, which do not express HDM-2 on their plasma membrane surfaces, are left entirely unaffected.
The peptide was developed at SUNY Downstate Medical Center in Brooklyn by a research group led by Matthew R. Pincus, with key contributions from Josef Michl (deceased 2022), Ehsan Sarafraz-Yazdi, and Wilbur Bowne. Over approximately 25 years of publications, this group has demonstrated PNC-27 activity against pancreatic cancer, breast cancer, melanoma, leukemia, ovarian cancer, lung cancer, colon cancer, sarcoma, and cervical cancer — in virtually every case killing cancer cells while leaving corresponding normal cells unharmed.[1]
The science is internally coherent, structurally grounded (NMR solution structure confirms p53-mimetic HDM-2 binding geometry), and supported by a body of in vitro and limited animal evidence. The concept of membrane HDM-2 as a selective cancer cell marker has received independent validation from a 2019 Nature Leukemia paper by a separate group targeting membrane HDM-2 in AML.[11]
| At a glance | |
|---|---|
| Full name | PNC-27 |
| Type | 32-residue chimeric peptide; rationally designed |
| Composition | p53 HDM-2-binding domain (residues 12–26) + C-terminal penetratin sequence from Antennapedia homeodomain |
| Related peptide | PNC-28 (shorter analogue, residues 17–26 of p53) |
| Mechanism | “Poptosis” — membrane HDM-2-targeted transmembrane pore formation → rapid tumour cell necrosis |
| Target | HDM-2 expressed on cancer cell plasma membrane |
| p53 dependence | None — kills p53-null cancer cells (K562 leukemia, MDA-MB-157 breast cancer) |
| Normal cell safety | No effect on multiple normal cell lines across all published studies (in vitro) |
| Cancer types studied | Pancreatic, breast, melanoma, leukemia, ovarian, colon, lung, cervical, sarcoma |
| Human clinical trials | ❌ None |
| FDA status | ❌ Not approved; no IND filed |
The Biological Rationale: Why Membrane HDM-2?
The p53–MDM2 axis
The p53 tumour suppressor protein is kept at low levels in normal cells by MDM2/HDM-2, which binds to p53’s transactivation domain, ubiquitinates it, and targets it for proteasomal degradation. In cancer, this system is deranged in two major ways: either p53 is mutated and non-functional (~50% of all cancers), or MDM2/HDM-2 is overexpressed, suppressing wild-type p53 and preventing the protective apoptotic response. Classical approaches (nutlin-3, RG7112, and others) block the intracellular p53–MDM2 interaction — but these require functional p53 and act through intracellular signalling.
The membrane HDM-2 observation
The key insight underlying PNC-27 is different: HDM-2 is not only expressed intracellularly — cancer cells express HDM-2 on the outer surface of their plasma membranes, a phenomenon not observed in normal cells. PNC-27 has no effect on untransformed cells which do not express HDM-2 in their cell membranes, and induces tumour cell necrosis of a wide range of cancer cells including solid tissue tumours and haematopoietic cancer cells, but has no effect on corresponding normal cells.[4] Why do cancer cells express HDM-2 on their membranes? The mechanism is not fully characterised, but appears related to the altered intracellular trafficking that accompanies oncogenic transformation.
This observation has received at least partial independent validation: a 2019 paper in Nature Leukemia (Wang et al.), from a separate research group not affiliated with the Pincus/Michl group, reported that targeting cell membrane HDM-2 is a novel therapeutic approach for acute myeloid leukemia — confirming that membrane HDM-2 exists on AML cells and can be exploited therapeutically.[11]
Structure and Design
The chimeric architecture
PNC-27 was designed through rational molecular modelling by Kanovsky et al. (2001, PNAS).[1] It has two functional domains. Domain 1 (N-terminal): amino acid residues 12–26 of the p53 transactivation domain — the region of p53 that natively binds to the N-terminal domain of MDM2/HDM-2. The key binding residues Leu22, Trp23, and Leu26 form the hydrophobic face of the helix that inserts into the MDM2 binding cleft. Domain 2 (C-terminal): a sequence from the Antennapedia homeodomain, a well-characterised cell-penetrating peptide. This domain enables membrane interaction and the physical insertion necessary for pore formation. The combined 32-residue peptide is not simply a delivery vehicle — the penetratin sequence actively participates in the membrane disruption that follows HDM-2 binding.
NMR structure confirmation
The solution structure of PNC-27 was determined by 2D-NMR and found that p53 residues 17–26 were superimposable on the X-ray crystal structure of the 15–29 p53 peptide bound to HDM-2 residues 1–109. Critical binding residues including Leu22, Trp23, and Leu26 interact with HDM-2 residues Ile99, Leu54, Ile61, and Met62 — and these critical interactions were preserved in both structures.[3] This structural confirmation is important: PNC-27 does not merely contain the correct sequence to bind HDM-2 — it adopts the correct three-dimensional conformation to do so.
Mechanism: Poptosis
The term “poptosis” (peptide-induced transmembrane pore formation) was coined by the Pincus group to distinguish PNC-27’s mechanism of cancer cell death from apoptosis. Apoptosis is internally programmed cell death proceeding through caspase activation, requiring the cell’s internal machinery to be functional — blocked in many cancers by p53 mutation, Bcl-2 overexpression, and caspase defects. Poptosis is external membrane rupture through pore formation → explosive release of intracellular contents → necrosis. It does not require functional p53 or intact apoptotic signalling, depending only on membrane HDM-2 expression.[6]
The four-step model: PNC-27 binds to HDM-2 associated with the membranes of susceptible tumour cells → PNC-27–HDM-2 complexes form ring-shaped transmembrane pore structures → the pores allow uncontrolled ion and molecule flux → rapid loss of membrane integrity leads to explosive release of intracellular contents and tumour cell death.
Immuno-scanning electron microscopy evidence
Immuno-scanning electron microscopy of cancer cells treated with PNC-27 and decorated with an anti-PNC-27 antibody coupled to 6 nm gold particles and an anti-HDM-2 antibody linked to 15 nm gold particles found multiple 6 nm- and 15 nm-labelled gold particles in approximately 1:1 ratios in layered ring-shaped structures in the pores near the cell surface, suggesting that PNC-27–HDM-2 1:1 complexes are important to the pore structure.[5] This is direct visualisation of PNC-27 and HDM-2 co-localised in ring-shaped pore structures at the cancer cell surface, consistent with the binding model.
Mitochondrial disruption: an additional mechanism
The 2024 Krzesaj et al. paper in Annals of Clinical and Laboratory Science added a second mechanistic dimension: PNC-27 not only disrupts the plasma membrane but also specifically disrupts cancer cell mitochondria through a similar pore-forming mechanism, while leaving normal cell mitochondria unaffected. This expands the mechanism from purely membranolytic to include organelle-level targeting.[8]
The monoclonal antibody blocking experiment
Incubating MIA-PaCa-2 human pancreatic carcinoma cells with PNC-27 in the presence of a monoclonal antibody directed against the amino-terminal p53-binding site of HDM-2 (residues 1–109) blocked PNC-27-induced tumour cell necrosis. The negative control immune serum did not block killing. This directly demonstrates that PNC-27’s cytotoxic activity requires binding to the specific p53-binding domain of membrane-expressed HDM-2 — confirming that the mechanism is exactly as proposed.
Cancer Types Studied
Pancreatic cancer (most studied)
MIA-PaCa-2 human pancreatic carcinoma cells are the primary model system, with dose-dependent necrosis, LDH release, pore formation by TEM, and HDM-2 membrane co-localisation by confocal microscopy confirmed. Normal rat pancreatic acinar cells (BMRPA1) remained fully viable at the same concentrations. PNC-28 blocked pancreatic cancer cell growth in vivo in the Michl et al. 2006 International Journal of Cancer paper — the most important animal model result in the series.[10]
Breast cancer
Do et al. (Oncogene, 2003): PNC-27 induced preferential necrosis in MCF-7 (wild-type p53), MDA-MB-468 (mutant p53), and MDA-MB-157 (p53-null) breast cancer cells. Normal MCF-10-2A breast cells were unaffected.[2] The p53-null killing confirmed p53-independence — the peptide kills regardless of p53 status because it acts at the membrane, not intracellularly.
Leukemia
K562 cells (chronic myelogenous leukemia, p53-null) showed near-complete killing. AML cell lines expressing membrane HDM-2 were killed. Critically, normal human haematopoietic stem cells and normal murine leukocytes were unaffected — suggesting no bone marrow suppression at therapeutic concentrations, a finding that would distinguish PNC-27 from conventional chemotherapy if confirmed in vivo.
Ovarian cancer
Sarafraz-Yazdi et al. (2015): ex vivo testing of PNC-27 against patient-derived epithelial ovarian cancer cells obtained from patients under IRB protocol showed killing, with a dose-response relationship correlating with membrane HDM-2 expression levels — among the most clinically relevant experiments in the series.
Cervical cancer (2025)
Three different cervical cancer cell lines (HTB-35, SW756, HeLa) were killed by PNC-27 with IC50 values among the lowest found for a wide variety of cancers studied. PNC-27 had no effect on the viability or growth of PCS-480-011 primary normal cervical epithelial cells. A 2025 paper additionally found that ketone bodies (lithium acetoacetate) enhanced PNC-27’s killing effect — a potentially synergistic combination for future study.[9]
Normal Cell Safety: The Key Claim
| Normal cell type | Tested against cancer counterpart | Finding |
|---|---|---|
| Normal rat pancreatic acinar cells (BMRPA1) | Pancreatic carcinoma (TUC-3) | Normal cells unaffected; cancer cells killed |
| Normal breast cells (MCF-10-2A) | MCF-7, MDA-MB-157, MDA-MB-468 | Normal cells unaffected |
| Normal cervical epithelial cells (PCS-480-011) | HTB-35, SW756, HeLa | Normal cells unaffected |
| Normal murine leukocytes | K562 leukemia | Normal cells unaffected |
| Normal human haematopoietic stem cells | AML cells | Normal stem cells unaffected |
The mechanistic basis for this selectivity is the differential expression of HDM-2 on the plasma membrane: expressed on cancer cell surfaces, absent or undetectable on normal cell surfaces. ⚠️ Important caveat: all normal cell safety testing has been performed in vitro. Systemic administration in a whole animal or human would expose PNC-27 to far more cell types, tissue environments, and physiological variables than a cell culture dish. Normal cells under inflammatory stress, cells with high rates of division, and cells in microenvironments that might induce transient HDM-2 membrane expression are not accounted for by in vitro data. The extraordinary selectivity claims require in vivo validation.
Evidence Summary
| Domain | Model | Finding | Evidence quality |
|---|---|---|---|
| NMR structure confirmation [3] | In silico / in vitro | PNC-27 adopts p53-mimetic binding conformation for HDM-2 | Strong structural |
| Plasma membrane pore formation [5] | TEM / immuno-SEM | Ring-shaped PNC-27–HDM-2 1:1 complexes in cancer cell membrane pores | Strong mechanistic |
| Pancreatic cancer cell killing | MIA-PaCa-2 in vitro | Dose-dependent necrosis with LDH release | Moderate (in vitro) |
| Breast cancer killing (p53-null) | MDA-MB-157 in vitro | Killing confirmed; p53-independence established | Moderate (in vitro) |
| Ovarian cancer (patient-derived) | Ex vivo primary cells | Killed by PNC-27 under IRB protocol | Moderate (ex vivo) |
| Normal cell safety (multiple types) | Multiple normal cell lines | Consistently unaffected across 5+ normal cell types | Moderate (in vitro only) |
| Anti-HDM-2 antibody blocking | MIA-PaCa-2 | Anti-HDM-2 antibody blocks killing; confirms mechanism specificity | Strong mechanistic |
| In vivo tumour suppression [10] | Rodent model | PNC-28 blocked pancreatic tumour growth; no off-target effects | Limited (one model) |
| Human clinical trials | N/A | None | ❌ No data |
Limitations: The 25-Year Gap
PNC-27 has been in active publication since the Kanovsky et al. 2001 PNAS paper. Twenty-five years is an extraordinary amount of time for a compound with the claimed properties — selective killing of cancer cells of virtually every type, no normal cell toxicity, in vivo tumour eradication — to remain at the preclinical stage with no IND application and no human trial.
All research originates from one primary group. No independent research group has replicated the core findings. For a compound claiming universal cancer cell killing with zero normal cell toxicity, independent replication is not optional — it is the minimum standard before any serious development investment. The in vitro concentrations are clinically problematic. Effective concentrations in most studies range from 10–500 µg/mL. Achieving and maintaining these concentrations systemically in a human without toxicity would require detailed pharmacokinetic work that has not been published. Systemic delivery challenges are unresolved. Peptides are degraded by serum proteases, renally cleared, and may not penetrate solid tumour stroma at therapeutic concentrations. Some cancers may have minimal membrane HDM-2. Low-grade prostate cancer, clear cell renal carcinoma, papillary thyroid carcinoma, and certain indolent lymphomas may exhibit minimal or absent HDM-2 expression, especially on the cell membrane; PNC-27 would not be expected to work for these tumour types.
Common Misconceptions
“PNC-27 has been proven to cure cancer.”
PNC-27 has demonstrated selective cancer cell killing in vitro and limited in vivo activity with PNC-28 in one animal model. No human data exists. The word “cure” does not apply.
“Because it targets HDM-2 on the cell surface, it will work for all cancers.”
HDM-2 membrane expression varies by cancer type, grade, and individual tumour. Some cancers express minimal membrane HDM-2. Tumour heterogeneity within a cancer type means responses would likely vary. Universal activity is not a justified inference from the current data.
“The normal cell safety data means it has no side effects in humans.”
Normal cell safety in vitro means nothing about systemic toxicity in a whole organism. PNC-27 at high concentrations encountering red blood cells, vascular endothelium, rapidly dividing cells, or immune cells in a physiological setting has not been tested in a systematic in vivo safety programme.
“PNC-27 works through apoptosis like other anticancer drugs.”
PNC-27 induces necrosis through pore formation (poptosis), not apoptosis. This is significant because it bypasses the apoptotic signalling defects (p53 mutation, Bcl-2 overexpression, caspase deficiency) that cause resistance to most conventional chemotherapy and targeted agents.
Frequently Asked Questions
Why hasn’t PNC-27 entered clinical trials after 25 years?
The most likely contributors: inadequate funding for IND-enabling pharmaceutical development studies; lack of a commercial partner willing to invest in clinical development based solely on one group’s preclinical data without independent replication; pharmacokinetic challenges that make clinical translation difficult; and the challenge of establishing which patients should be selected based on membrane (not intracellular) HDM-2 expression status. The absence of a clinical trial is not evidence that PNC-27 doesn’t work — it is evidence that the path from promising preclinical data to clinical trial has not been successfully navigated.
Does PNC-27 affect tumour suppressor p53 directly?
No. PNC-27 mimics a fragment of p53 to bind HDM-2 at the cell membrane. It does not restore p53 function, does not activate p53 signalling, and does not require p53 to be present or functional for its cytotoxic effect. This distinguishes it fundamentally from MDM2 inhibitors like nutlin-3, which require wild-type p53 to work.
What about PNC-27 and pancreatic cancer specifically?
Pancreatic ductal adenocarcinoma is one of the deadliest cancers, with almost universal resistance to chemotherapy partly due to KRAS mutation and p53 inactivation. PNC-27’s mechanism is p53-independent and acts at the membrane — theoretically bypassing these resistance mechanisms. The pancreatic cancer data is the most developed in the PNC-27 literature. However, “most developed” in this context still means in vitro, one PNC-28 animal study, and no human data.[10]
Key Takeaways
- The mechanism is structurally grounded and conceptually novel. NMR confirms the p53-mimetic binding conformation.[3] Immuno-SEM directly visualises pore formation.[5] The anti-HDM-2 blocking antibody experiment confirms mechanistic specificity. Poptosis is mechanistically distinct from any approved cancer drug’s mode of action.
- The selectivity claim is supported by consistent in vitro data across multiple cancer types and multiple normal cell types. The mechanistic basis (differential membrane HDM-2 expression) is biologically plausible and has received independent target validation in AML.[11]
- ⚠️ The entire evidence base comes from one primary research group. Independent replication by unaffiliated laboratories is essential before PNC-27 can be considered for clinical development.
- ⚠️ No human data exists. Twenty-five years of publication without an IND application is a significant gap. The in vitro concentrations and systemic delivery challenges are unresolved.
- ⚠️ Patients with cancer should pursue evidence-based treatment. PNC-27 is not a treatment option — it is a research compound at an early preclinical stage. Established therapies for each cancer type, reviewed with an oncologist familiar with the individual patient’s staging, genomics, and treatment history, represent the appropriate standard of care.
References
Founding Papers
- Kanovsky M, Raffo A, Drew L, Rosal R, Do T, et al. Peptides from the amino terminal MDM-2 binding domain of p53, designed from conformational analysis, are selectively cytotoxic to transformed cells. Proceedings of the National Academy of Sciences USA. 2001;98:12438–12443.
- Do TN, Rosal RV, Drew L, Raffo AJ, et al. Preferential induction of necrosis in human breast cancer cells by a p53 peptide derived from the MDM-2 binding site. Oncogene. 2003;22:1431–1444.
Structural and Mechanistic
- Rosal R, Pincus MR, Brandt-Rauf PW, et al. NMR solution structure of a peptide from the MDM-2 binding domain of the p53 protein that is selectively cytotoxic to cancer cells. Biochemistry. 2004;43:1754–1761.
- Sarafraz-Yazdi E, Bowne WB, Adler V, et al. Anticancer peptide PNC-27 adopts an HDM-2-binding conformation and kills cancer cells by binding to HDM-2 in their membranes. Proceedings of the National Academy of Sciences USA. 2010;107:1918–1923. PMC2836618
- Sarafraz-Yazdi E, Mumin S, Cheung D, et al. PNC-27, a chimeric p53-penetratin peptide binds to HDM-2 in a p53 peptide-like structure, induces selective membrane-pore formation and leads to cancer cell lysis. Biomedicines. 2022;10:945.
Reviews: Poptosis Mechanism
- Pincus MR, Bowne WB, Sarafraz-Yazdi E. Poptosis: a novel mechanism for the selective killing of cancer cells. Clinical Oncology. 2023;7(6):1–13.
- Pincus MR, Silberstein M, Zohar N, Sarafraz-Yazdi E, Bowne WB. Poptosis or peptide-induced transmembrane pore formation: a novel way to kill cancer cells without affecting normal cells. Biomedicines. 2024;12(6):1144. PMC11201261
Recent Studies
- Krzesaj P, Adler V, Feinman RD, Miller A, et al. Anti-cancer peptide PNC-27 kills cancer cells by unique interactions with plasma membrane-bound HDM-2 and with mitochondrial membranes causing mitochondrial disruption. Annals of Clinical and Laboratory Science. 2024;54(2):137–148.
- Miller AI, et al. PNC-27 kills cervical cancer cells but not untransformed cervical cells, an effect that is enhanced by ketone bodies. Medical Research Archives. 2025;13(5). doi: 10.18103/mra.v13i5.6471
In Vivo Evidence
- Michl J, Scharf B, Schmidt A, Hannan R, et al. PNC-28, a p53 peptide that is cytotoxic to cancer cells, blocks pancreatic cancer cell growth in vivo. International Journal of Cancer. 2006;119:1577–1585.
Independent Target Validation
- Wang H, Zhao D, Nguyen LX, et al. Targeting cell membrane HDM2: a novel therapeutic approach for acute myeloid leukemia. Leukemia. 2019;34:75–86.
Key Investigators
- Matthew R. Pincus, MD, PhD — Department of Pathology, SUNY Downstate Medical Center, Brooklyn, NY; primary author and PI of the PNC-27 programme from inception through present.
- Josef Michl, MD, PhD (1943–2022) — Department of Pathology, SUNY Downstate; co-founder of the PNC-27 programme and co-author of the first in vivo efficacy paper (PNC-28, 2006).
- Ehsan Sarafraz-Yazdi, PhD — NomoCan Pharmaceuticals; co-lead author on the foundational 2010 PNAS structural paper; continuing investigator.
- Wilbur B. Bowne, MD — Department of Surgery, Thomas Jefferson University, Philadelphia; surgical oncologist and co-investigator; lead on the pancreatic cancer and ovarian cancer translational studies.
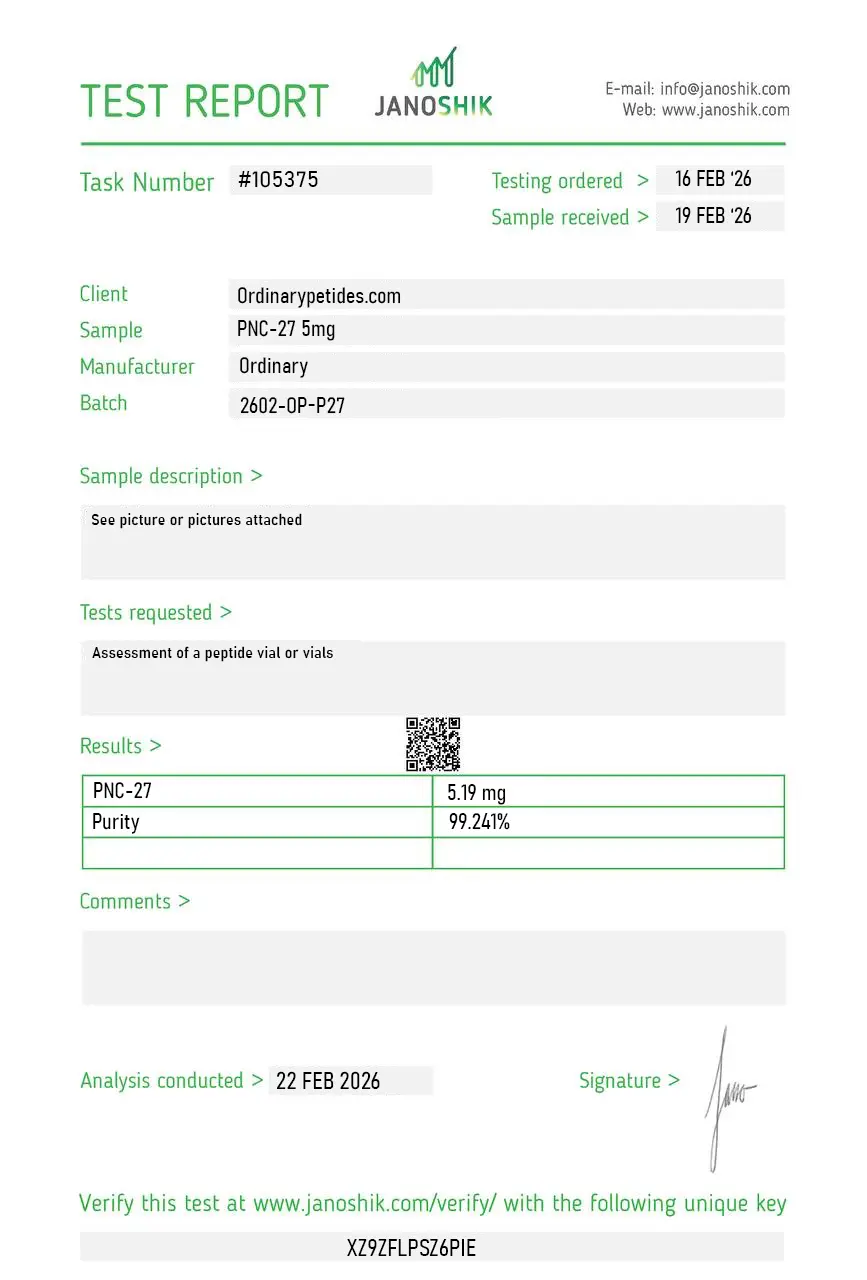
Certificate of Analysis
An independent test report is available for PNC-27 5mg. This report provides batch-level documentation and analytical verification information for research reference.
Based on 1 reviews
5.0
The membrane-disruption mechanism is what we study, not selectivity-as-therapy — PNC-27 is a membrane-perturbing peptide construct that produces necrotic cell death through bilayer compromise, not through clean apoptotic signaling. For probing membrane-active peptide cytotoxicity as a class — alongside other cationic amphipathic cytolytic peptides — PNC-27 is a defined construct example, and the necrotic death modality is the experimental object rather than a therapeutic feature. The material produced consistent membrane-permeabilization signatures in our cell-line assays.
Most correctly, it is a research anticancer peptide. There are no reliable grounds to regard it as an approved drug.
To use a p53-derived sequence to bind HDM-2/MDM2 and, according to the main publications, cause selective damage to tumor-cell membranes.
Based on the available reliable literature, that cannot be claimed. There is no convincing clinical basis in humans.
Yes. There are data from cell models, ex vivo tumor samples, AML, and paclitaxel-combination models. But this is still not proof of clinical benefit.
PNC-27 is longer and contains p53 residues 12–26, whereas PNC-28 contains residues 17–26; both peptides are linked to penetratin.
Because the molecule simultaneously:
- has a real scientific history and interesting preclinical work,
- does not have convincing clinical validation,
- has appeared in problematic commercial promotion.
This is almost the perfect recipe for myths to emerge
PNC-27 is a synthetic 32-amino acid chimeric peptide designed as a selective anticancer agent. It was created in 2000 using computational methods at SUNY Downstate Medical Center by a research group led by Pincus, Michl, and colleagues. It is constructed from two distinct functional domains fused together — the HDM-2 binding domain corresponding to amino acids 12 through 26 of the tumor suppressor protein p53, combined on its C-terminus with a cell-penetrating peptide (CPP) leader sequence derived from penetratin. The rationale was to create a molecule that could selectively home to cancer cells and destroy them through a mechanism fundamentally different from conventional chemotherapy. PNC-27 has no FDA approval, has not completed Phase 3 clinical trials, and is not commercially available as an approved therapeutic in the United States.
PNC-27's mechanism, termed "poptosis" (peptide-induced transmembrane pore formation) by its developers, is unlike any existing approved cancer therapy and genuinely distinct from most peptides in this series. The key insight from published research is that cancer cells — but not normal untransformed cells — express significant levels of HDM-2 (human double minute 2, also called MDM-2) protein in their plasma membranes. Normal cells contain HDM-2 only in the nucleus and cytoplasm, not the cell membrane. PNC-27 is structurally designed to adopt the same three-dimensional conformation as p53 residues 12–26 when bound to HDM-2, allowing it to bind with high affinity to the membrane-bound HDM-2 that is uniquely present on cancer cell surfaces. Once bound, the penetratin CPP domain inserts into the lipid bilayer and drives the formation of transmembrane pores — ring-shaped structures confirmed by immuno-scanning electron microscopy decorated with both anti-PNC-27 and anti-HDM-2 gold-labeled antibodies in approximately 1:1 ratios. These pores cause rapid leakage of cellular contents — lactate dehydrogenase release occurs within minutes — leading to tumor cell necrosis rather than apoptosis. Because untransformed normal cells do not express HDM-2 in their membranes, PNC-27 has no mechanism to bind them and produces no pores in normal cell membranes.
This is one of the most clinically important features of PNC-27's mechanism. Approximately 50% of all human cancers harbor p53 mutations, and many cancer therapies that work by restoring p53 tumor suppressor function — including small molecule MDM-2 inhibitors — are ineffective in p53-null or p53-mutant cancers. PNC-27 has been shown to kill cancer cell lines that are p53 homozygously deleted — including K562 leukemia cells and MDA-MB-157 breast cancer cells with null p53 — because its mechanism operates entirely through membrane-bound HDM-2 and does not require functional p53 activity. This means it could theoretically address cancers that are resistant to p53-pathway-dependent therapies.
Published peer-reviewed research has documented PNC-27 cytotoxicity against a broad range of cancer cell lines and primary human cancer specimens — pancreatic cancer, breast cancer (including mutant p53, overexpressed wild-type p53, and p53-null variants), colon adenocarcinoma, cervical cancer, melanoma, non-small cell lung cancer, osteosarcoma, leukemia, ovarian cancer, and rat k-ras-transformed pancreatic cancer. In every study untransformed normal cell controls — including primary human fibroblasts, normal cervical epithelial cells, and human umbilical cord hematopoietic stem cells — showed no toxicity. In vivo studies in nude mice showed PNC-28 (a related analog covering p53 residues 17–26) eradicated pancreatic cancer xenografts with no off-target effects. A 2025 publication documented PNC-27's efficacy against three cervical cancer cell lines with IC50 values among the lowest the research group had found across a wide variety of cancers.
This requires careful and honest assessment. The research group's publications are genuine, peer-reviewed, and published in legitimate journals including PNAS. However the claim appearing in some online sources that "clinical trials were successful and the drug is currently in use outside of the United States" is not supported by evidence in the published peer-reviewed literature or major clinical trial registries as of April 2026. No Phase 3 trial data has been published. The compound remains in preclinical and early exploratory research stages. It is not approved by the FDA, EMA, or any major regulatory authority. It is not legally available as a therapeutic agent through any legitimate pharmaceutical channel. Anyone claiming access to approved PNC-27 treatment outside the United States should be approached with significant skepticism.
In the published preclinical research PNC-27 has shown a remarkably selective safety profile — it kills cancer cells but not normal cells across multiple independent experiments and cell types. No significant off-target effects were observed in vivo in the mouse studies. No serious side effects were documented in the very limited human exposure data referenced in student publications. However the absence of formal Phase 1 dose-escalation human safety data means the human safety profile — particularly at therapeutic doses, with systemic administration routes and in patients with comorbidities — is genuinely unknown.
Everyone should not use PNC-27 outside of a formally approved clinical trial. There is no legitimate therapeutic access channel for this compound. Anyone offering PNC-27 as a cancer treatment outside of a registered clinical trial is operating outside the law and outside the science. Patients with cancer should discuss all available treatments — including clinical trial participation — with a qualified oncologist. The mechanistic science behind PNC-27 is genuinely interesting and the preclinical data is encouraging, but that does not make it a treatment option. Patients should be especially cautious of cancer treatment claims made outside the peer-reviewed literature and regulatory approval framework.