PE-22-28
For in vitro testing and laboratory use only. Not for human or animal consumption. Bodily introduction is illegal. Handle only by licensed professionals. Not a drug, food, or cosmetic. Educational use only.
PE-22-28: Tiny Sequence, Big Research Curiosity
PE-22-28 is a short 7-amino-acid peptide created as a shortened analog of spadin, and the interest in it did not arise out of nowhere: in the research context, it showed very high activity toward TREK-1, a potassium channel that has long attracted attention in the neurobiology of stress, mood, and plasticity. In published preclinical studies, PE-22-28 appeared as a more potent variant compared with spadin in electrophysiological systems, and in animal models it was associated with antidepressant-like effects, as well as with changes that researchers linked to neurogenesis and synaptogenesis.
The story did not stop there: as part of ongoing study, it was also examined in post-stroke models and in the context of β-cell survival, which added further scientific intrigue to the molecule. But the whole honest essence is that PE-22-28 still exists mainly in the world of cells, channels, and animal models, not in the zone of confirmed clinical benefit in humans.
That is why it is compelling not as a "ready-made peptide for the brain," but as a carefully constructed research tool with strong preclinical logic and a still unresolved question of real-world translation. For a client, this is exactly the kind of case where the interest comes not from hype, but from the sense that behind a tiny molecule stands a big and genuinely intelligent scientific story.
PE-22-28: A Scientific Review
Based on peer-reviewed literature — see References. Last updated: April 2026.
The Short Version
PE-22-28 is a 7-amino acid synthetic peptide that blocks the TREK-1 potassium channel — a mechanistic target for depression that has nothing to do with serotonin, noradrenaline, or dopamine. It represents the latest iteration of a 15-year research programme at CNRS Nice, France, aimed at developing a new generation of antidepressants through an entirely different pharmacological approach than any currently approved drug.
The story requires understanding three things in sequence. In 2006, Heurteaux and colleagues published in Nature Neuroscience that mice lacking the TREK-1 potassium channel were spontaneously resistant to depression, behaving as if chronically treated with antidepressants, with elevated serotonin neuron firing and increased neurogenesis — without any pharmacological intervention.[1] In 2010, Mazella et al. published in PLOS Biology that a naturally occurring 17-amino acid peptide called spadin — released from the protein sortilin during normal processing — blocks TREK-1 and produces antidepressant effects in mice within 4 days (vs. 21 days for fluoxetine).[2] In 2017, Djillani et al. identified PE-22-28 as a 7-amino acid fragment with dramatically better TREK-1 affinity (IC50 = 0.12 nM vs. 40–60 nM for spadin — a ~330–500-fold improvement) and antidepressant effects in mice at doses 25-fold lower than spadin.[3]
The entire research programme comes from one laboratory group. No independent human pharmacological data exists. The mechanism is biologically compelling. The translational gap to humans is not yet bridged.
| At a glance | |
|---|---|
| Full name | PE-22-28 (also: mini-spadin, shortened spadin analogue) |
| Sequence | Gly-Gln-Pro-Pro-Ser-Thr-Asn-Lys (positions 22–28 of sortilin propeptide PE) |
| Length | 7 amino acids (heptapeptide) |
| Molecular weight | ~727 Da |
| Primary target | TREK-1 (TWIK-Related Potassium Channel 1; KCNK2) — K2P family |
| Mechanism | Selective TREK-1 channel blocker → increased serotonin neuron firing in dorsal raphe nucleus |
| IC50 for TREK-1 | 0.12 nM (vs. spadin: 40–60 nM; vs. fluoxetine: µM range) |
| Origin | Derived from spadin (PE 12-28), which is derived from the propeptide (PE) of sortilin (neurotensin receptor 3) |
| Discovery group | CNRS IPMC Nice, France — Jean Mazella, Catherine Heurteaux, Marc Borsotto, Abdulhamid Djillani |
| Human clinical trials | ❌ None |
| FDA/regulatory status | ❌ Not approved; research chemical only |
The Biology: TREK-1 and Depression
What are K2P potassium channels?
Two-pore domain potassium (K2P) channels are background potassium channels that are constitutively active under resting conditions, unlike voltage-gated or ligand-gated channels. K2P channels stabilise the resting membrane potential by providing a persistent “leak” of potassium ions out of the cell — keeping the membrane hyperpolarised and reducing neuronal excitability. TREK-1 (KCNK2) is the most extensively studied K2P channel and is highly expressed throughout the brain, including in the prefrontal cortex, hippocampus, amygdala, nucleus accumbens, and — critically — the dorsal raphe nucleus (DRN), which is the primary source of serotonergic projections to the forebrain. TREK-1 is also expressed in heart, smooth muscle cells, endocrine pancreas, and prostate.[4]
The 2006 Nature Neuroscience turning point
The behaviour of kcnk2&sup-;/&sup-; mice was similar to those treated acutely or chronically with classical antidepressants like fluoxetine. These mice show an increase in serotonin (5-HT) neurotransmission in the DRN neurons, and deletion of TREK-1 increases neurogenesis. The finding was significant for three reasons: it identified a novel molecular target for depression entirely outside the monoamine system; it demonstrated a depression-resistant phenotype across five different behavioural tests; and it explained why SSRIs might block TREK-1 as a secondary mechanism — at clinical concentrations, SSRIs inhibit TREK-1, providing a potential explanation for their antidepressant activity beyond serotonin reuptake inhibition.[1]
The Sortilin–PE–Spadin–PE-22-28 Derivation Chain
Sortilin and the propeptide PE
Sortilin is a transmembrane receptor with roles in intracellular protein sorting, neurotrophin signalling (co-receptor for pro-BDNF and pro-NGF), and lysosomal targeting. Sortilin is synthesised as prosortilin, which is cleaved in the trans-Golgi compartment by the proprotein convertase furin to generate mature sortilin and release a 44-amino acid propeptide (PE). When COS-7 cells expressing TREK-1 are treated with PE, arachidonic acid-induced channel activation is reduced by 70–80%. Crucially, neurotensin (the natural sortilin ligand) does not produce this TREK-1 inhibitory effect — it is specific to PE and its derivatives.[6]
Spadin: PE positions 12–28
Spadin (PE 12-28) was rationally designed by taking the core TREK-1-binding sequence of PE (positions 17–28) and adding an N-terminal stabilising sequence. This 17-amino acid peptide bound TREK-1 with IC50 of 40–70 nM. The 2010 PLOS Biology paper (Mazella et al.) validated spadin in five mouse behavioural models of depression — FST, TST, NSF, LHT, and corticosterone-induced model — producing antidepressant-like effects within 4 days and increasing hippocampal neurogenesis, without cardiac effects, sleep disruption, or seizure provocation. Spadin’s limitation: in vivo antidepressant effects disappeared 7 hours after administration, making it a poor drug candidate for chronic depression treatment.[2]
PE-22-28: the 7-amino acid optimised fragment
From the study of spadin blood degradation products, Djillani et al. designed a 7-amino acid peptide, PE-22-28, corresponding to positions 22–28 of the original 44-amino acid PE — the minimal sequence necessary for high-potency TREK-1 inhibition. In vitro studies on hTREK-1/HEK cells by patch-clamp technique showed that PE-22-28 displayed an IC50 of 0.12 nM vs. 40–60 nM for spadin — a ~330–500-fold improvement in TREK-1 affinity. Antidepressant effects in mice were achieved at doses ~25-fold lower than spadin.[3]
Mechanism of Action
TREK-1 channel blockade: potency and selectivity
PE-22-28 inhibits TREK-1 currents with IC50 = 0.12 nM using patch-clamp electrophysiology on human TREK-1-expressing HEK293 cells. Current blockade is specific to TREK-1, as TREK-2, TRAAK, TRESK, and TASK-1 channels are not inhibited. Critically, spadin-analogues including PE-22-28 are without effect on hERG channel activity — the cardiac safety ion channel whose inadvertent blockade underlies QT prolongation and potentially fatal arrhythmias.[3]
The serotonin connection: the dorsal raphe mechanism
TREK-1 channels in DRN serotonergic neurons keep them tonically hyperpolarised. By blocking TREK-1, PE-22-28 reduces this hyperpolarisation, increasing DRN 5-HT neuron firing rate and downstream serotonergic transmission. This is mechanistically analogous to — but pharmacologically distinct from — SSRIs: SSRIs block SERT → reuptake inhibition → more serotonin in the synapse; PE-22-28 blocks TREK-1 → removes K+ leak hyperpolarisation → increased DRN serotonergic neuron firing → more serotonin released presynaptically. At clinical concentrations, SSRIs themselves inhibit TREK-1 as a secondary pharmacological effect, suggesting the channel may contribute to existing SSRI efficacy.[1]
Neurogenesis and synaptogenesis
PE-22-28 and its analogue G/A-PE-22-28 increase hippocampal neurogenesis (measured by BrdU labelling) and upregulate PSD-95, a marker of synaptogenesis and dendritic spine maturation. These downstream effects parallel those of chronic antidepressant treatment and are consistent with the neurotrophic hypothesis of depression. PE-22-28 achieves them within 4 days, compared to 21 days for fluoxetine.
The Spadin Family: Comparison of Key Compounds
| Compound | AA | IC50 TREK-1 | In vivo stability | Relative potency vs. spadin |
|---|---|---|---|---|
| Spadin (PE 12-28) | 17 | 40–70 nM | ~7 hours | Reference |
| PE-22-28 [3] | 7 | 0.12 nM | Not specifically reported | ~330–500× more potent |
| G/A-PE-22-28 | 7 | 0.10 nM | ~21 hours | ~400× more potent |
| Biotin-G/A-PE-22-28 | 7 (modified) | 1.2 nM | ~23 hours | ~33× more potent |
G/A-PE-22-28 (glycine at position 22 replaced by alanine) is the best-performing analogue for combined potency and in vivo stability, and is the closest candidate to a drug development lead from this series.
Preclinical Evidence
All evidence for PE-22-28 is preclinical and originates from the CNRS IPMC Nice group.
Behavioural depression models
PE-22-28 at 3–4 µg/kg (intraperitoneally) significantly reduced immobility time in the Forced Swim Test (FST) after both acute (30 minutes post-injection) and 4-day sub-chronic treatment, compared to spadin at 100 µg/kg — a ~25-fold dose reduction consistent with the affinity improvement. PE-22-28 also significantly improved outcomes in the Learned Helplessness Test, reduced latency in the Novelty Suppressed Feeding test, and reversed the corticosterone-induced depressive phenotype in acute and sub-chronic treatments.[3]
Route flexibility
PE-22-28 analogues reduce immobility time in the FST after intraperitoneal, intravenous, and oral gavage administration — the last of which is particularly significant for drug development, suggesting some efficacy is retained after gut exposure, though formal oral bioavailability has not been characterised.
Post-stroke depression
TREK-1 is overexpressed following cerebral ischaemia, contributing to post-stroke depression, which is resistant to conventional antidepressants. Pietri et al. (2019, Neuropharmacology) showed first evidence that sortilin-derived peptides have protective effects on stroke recovery and post-stroke depression — a clinically important unmet need.[7]
Seizure safety — an important negative finding
Despite prior concern that TREK-1 blockade might increase seizure susceptibility (given TREK-1’s neuroprotective role in ischaemia models), neither spadin nor PE-22-28 exacerbates seizure activity. In fact, spadin-treated mice exhibit heightened resistance to generalised seizures, with PE-22-28 appearing to exhibit even more profound protective effects. This is a reassuring preclinical safety signal.
Evidence Summary
| Endpoint | Model | Finding | Evidence quality |
|---|---|---|---|
| TREK-1 inhibition (in vitro) [3] | hTREK-1/HEK cells, patch-clamp | IC50 = 0.12 nM; selective vs. TREK-2, TRAAK, TRESK, TASK-1, hERG | Strong in vitro |
| FST (acute and sub-chronic) | Mouse | Significant immobility reduction at 3.2–4.0 µg/kg | Moderate preclinical |
| Learned Helplessness Test | Mouse | Significant improvement | Moderate preclinical |
| Novelty Suppressed Feeding | Mouse | Significant reduction in anxiety/depression behaviour | Moderate preclinical |
| Hippocampal neurogenesis | Mouse | Increased BrdU+ cells; faster than fluoxetine | Moderate preclinical |
| DRN 5-HT firing | Mouse (electrophysiology) | Increased serotonergic neuron firing rate | Strong electrophysiological |
| Post-stroke depression [7] | Mouse stroke model | Protective effects | Early preclinical |
| Seizure safety | Mouse | No seizure provocation; improved seizure resistance | Reassuring preclinical |
| hERG cardiac safety | In vitro | ✅ No hERG inhibition | Reassuring in vitro |
| Human clinical trials | None | No data | N/A |
Limitations: One Lab, One Species, No Humans
All research originates from one laboratory group. Every peer-reviewed paper on spadin, PE-22-28, and their analogues comes from the CNRS IPMC Nice group. No independent laboratory has replicated the core in vitro pharmacology or in vivo behavioural findings. For a compound targeting a genuinely novel mechanism with commercially compelling properties, the absence of independent replication is a significant epistemic limitation.
All in vivo data is from rodent models. The translation from mouse depression models to human clinical efficacy has a poor track record in psychiatry. TREK-1 expression patterns, receptor density in the DRN, and the relative contribution of the K2P pathway to depression differ between mouse and human biology in ways not fully characterised.
No human data of any kind. Not pharmacokinetic, not pharmacodynamic, not safety. The step from mouse intraperitoneal injection at 3–4 µg/kg to human oral or SC administration at unknown doses requires extensive work that has not been published. The “rapid onset” claim — 4 days vs. 2–6 weeks for SSRIs — requires human validation before it can be acted upon clinically.
Why PE-22-28 Is Scientifically Compelling Despite Its Limitations
Endogenous origin and biological coherence: PE-22-28 is derived from the naturally occurring endogenous propeptide released during sortilin maturation. The sortilin–TREK-1 protein complex is biochemically demonstrated. PE-22-28 is an optimised fragment of a biologically real endogenous signal, not a designed synthetic entity acting on an arbitrary target.[6]
Genetic validation of the target: The kcnk2&sup-;/&sup-; mouse phenotype provides genetic proof-of-concept — TREK-1 deletion alone produces depression resistance — at a level of evidence independent of the Nice group’s own pharmacological work.[1]
Mechanism independent from monoamine theory: Depression’s biological basis remains poorly understood, and the dominant serotonin/noradrenaline hypothesis is inadequate to explain delayed SSRI onset or non-response in 30–40% of patients. TREK-1 represents a complementary mechanistic framework that could address cases where the monoamine system is not the primary driver.
The in vitro potency is extraordinary: IC50 = 0.12 nM for TREK-1, with selectivity demonstrated across five related K2P channels and no hERG activity. At this affinity, therapeutic channel blockade may be achievable at very low systemic concentrations, reducing off-target exposure.[3]
Common Misconceptions
“PE-22-28 blocks serotonin reuptake like SSRIs.”
PE-22-28 does not act on the serotonin transporter (SERT) at all. It acts on a potassium channel (TREK-1) that regulates the excitability of serotonergic neurons upstream. The outcome — increased serotonergic transmission — is mechanistically related to SSRIs’ effect, but the pharmacological target is completely different.
“The studies show it works in humans.”
All published efficacy data is in mice. The compound has never been tested in human pharmacological or clinical studies.
“The faster onset means it’s definitely better than SSRIs for humans.”
The 4-day vs. 21-day comparison is in mice, measured by FST immobility, translated to human depression duration and outcome criteria with multiple assumptions that have not been validated.
Frequently Asked Questions
What does “PE” stand for in PE-22-28?
PE is the 44-amino acid propeptide released when sortilin (neurotensin receptor 3) matures through furin cleavage in the trans-Golgi network. The “22-28” refers to positions 22 through 28 of this 44-amino acid propeptide sequence — the minimal active fragment for high-potency TREK-1 inhibition.
Is PE-22-28 the same as spadin?
No. Spadin is PE 12-28 (positions 12–28 of the propeptide) — a 17-amino acid peptide. PE-22-28 is a 7-amino acid fragment (positions 22–28 only), designed by analysing spadin’s degradation products in blood and identifying the minimal effective sequence. PE-22-28 is about 330–500-fold more potent than spadin in vitro.
Why haven’t clinical trials started yet if this was published in 2017?
The transition from academic preclinical research to IND-enabling studies requires significant pharmaceutical development work: formulation optimisation, GMP manufacture, formal toxicology package (rodent and non-rodent 28-day toxicology, genotoxicity, safety pharmacology), and pharmacokinetic characterisation in non-rodent species. The Nice group appears to have continued academic characterisation rather than entering the pharmaceutical development pipeline — a gap that often separates academic discovery from clinical translation, particularly for peptide-based drugs.
Key Takeaways
- PE-22-28 is pharmacologically exceptional for its target. An IC50 of 0.12 nM with demonstrated selectivity across five related channels and no hERG activity represents genuinely rigorous in vitro pharmacology. The compound is real and potent at its target.[3]
- The TREK-1/depression biology has strong independent genetic validation. The kcnk2&sup-;/&sup-; mouse phenotype from Heurteaux et al. (2006) establishes TREK-1 as a causal contributor to depression biology, independent of the Nice group’s own pharmacological work.[1]
- ⚠️ The entire in vivo evidence base comes from one research group. No independent laboratory has replicated the antidepressant behavioural effects of spadin or PE-22-28 in mice, let alone validated the mechanism in human tissue.
- ⚠️ No human data exists. This is an early-stage preclinical research compound. The clinical translation pathway has not been entered.
- The potential clinical differentiation is real and meaningful. A 4-day antidepressant onset with a non-monoamine mechanism, no hERG effects, no seizure provocation, and selectivity for TREK-1 over related channels would be a genuine advance in depression pharmacotherapy — if the mouse findings translate.
- ⚠️ For individuals with treatment-resistant depression: PE-22-28 is not a treatment option — it is a preclinical research compound. The appropriate path is working with a psychiatrist to explore approved alternatives including SNRIs, bupropion, mirtazapine, ketamine/esketamine (FDA-approved for treatment-resistant depression), and augmentation strategies.
References
TREK-1 as Depression Target
- Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thümmler S, et al. Deletion of TREK-1, a background potassium channel, results in a depression-resistant phenotype. Nature Neuroscience. 2006;9:1134–1141.
Discovery of Spadin
- Mazella J, Pétrault O, Lucas G, Deval E, et al. Spadin, a sortilin-derived peptide, targeting rodent TREK-1 channels: a new concept in the antidepressant drug design. PLOS Biology. 2010;8(4):e1000355. PMC2854129
PE-22-28 Key Paper
- Djillani A, Pietri M, Moreno S, Heurteaux C, Mazella J, Borsotto M. Shortened spadin analogs display better TREK-1 inhibition, in vivo stability and antidepressant activity. Frontiers in Pharmacology. 2017;8:643. PMC5601071
TREK-1 Biology Reviews
- Djillani A, Mazella J, Heurteaux C, Borsotto M. Role of TREK-1 in health and disease, focus on the central nervous system. Frontiers in Pharmacology. 2019;10:379. PMC6470294
- Borsotto M, Veyssière J, Moha Ou Maati H, Devader C, Mazella J, Heurteaux C. Targeting two-pore domain K+ channels TREK-1 and TASK-3 for the treatment of depression: a new therapeutic concept. British Journal of Pharmacology. 2015;172:771–784.
Sortilin and TREK-1
- Mazella J, Borsotto M, Heurteaux C. The involvement of sortilin/NTSR3 in depression as the progenitor of spadin and its role in the membrane expression of TREK-1. Frontiers in Pharmacology. 2018. PMC6331531
Post-Stroke Depression
- Pietri M, Djillani A, Mazella J, Borsotto M, Heurteaux C. First evidence of protective effects on stroke recovery and post-stroke depression induced by sortilin-derived peptides. Neuropharmacology. 2019;158:107715.
Key Investigators
- Jean Mazella, PhD — CNRS IPMC, Université Côte d’Azur, Valbonne, France; discoverer of spadin (2010); lead author on the propeptide-TREK-1 link; principal investigator of the sortilin-derived antidepressant programme.
- Catherine Heurteaux, PhD — CNRS IPMC Nice; lead author on the foundational 2006 Nature Neuroscience TREK-1 depression paper; co-investigator of the spadin/PE-22-28 programme.
- Abdulhamid Djillani, PhD — First author on the 2017 Frontiers in Pharmacology paper describing PE-22-28 and its analogues as lead compounds for clinical development.
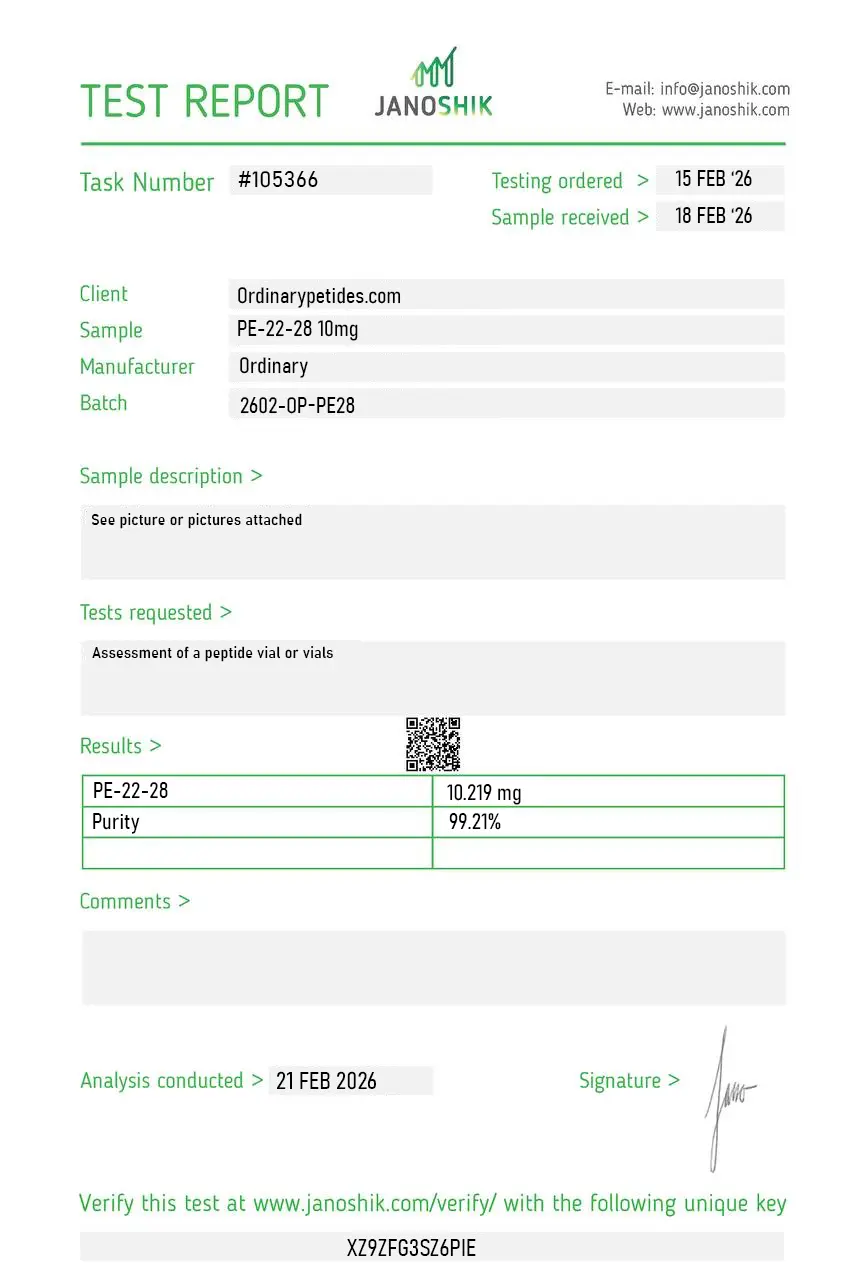
Certificate of Analysis
An independent test report is available for PE-22-28 10mg. This report provides batch-level documentation and analytical verification information for research reference.
Based on 1 reviews
5.0
We anchor a K2P-channel pharmacology series with PE-22-28 as the TREK-1-selective reference — PE-22-28 against the broader two-pore-domain blockers and against TREK-1-activating compounds, characterizing how the channel's open versus closed states drive different neuronal phenotypes. That selectivity across closely-related channels is the experimental object, and PE-22-28's reasonable selectivity is what makes it the practical tool for the TREK-1 arm. The material held that selectivity profile across our patch-clamp conditions.
No. Spadin is a longer peptide (PE 12-28), while PE-22-28 is its shortened active analog.
Based on the available literature — there are no grounds to claim that. It is more accurate to speak of a preclinical candidate.
Mainly, the discussion concerns biomarkers of sortilin-derived propeptide and the broader TREK-1/sortilin concept. There is still no convincing clinical base for PE-22-28 therapy itself in humans.
Because deletion of TREK-1 in mice produced a depression-resistant phenotype, which made the channel a new potential target in psychopharmacology.
This is a marketing category, not a strictly scientific one. Based on the published data, it is more accurate to speak of preclinical effects on neuroplasticity and behavioral models, not a proven "nootropic effect" in humans.
A complete human side-effect profile has not been established. The preclinical data for spadin and its analogs look encouraging, but that is not enough for confident conclusions about safety in humans.
In preclinical models — yes: higher activity on TREK-1 and improved duration of effect for a number of analogs. But the clinical significance of these advantages has not yet been proven.
PE-22-28 is a synthetic seven-amino acid peptide derived from spadin — itself a naturally occurring peptide fragment cleaved from the propeptide region of sortilin, a multifunctional receptor expressed in the brain and periphery. Spadin was identified in 2011 by French researchers as an endogenous TREK-1 potassium channel antagonist with antidepressant properties in animal models. PE-22-28 was then developed as a more potent, more stable, shorter analog of spadin — designed by the same French research group to address spadin's short in vivo half-life of approximately 7 hours. It represents one of a novel class of antidepressant research compounds targeting a completely different molecular mechanism from all currently approved antidepressants. It has no FDA approval, no completed human clinical trials, and is available only as a research compound.
TREK-1 (TWIK-Related K+ Channel-1) is a two-pore domain background potassium channel expressed widely in the central nervous system — particularly in regions relevant to mood regulation including the cortex, hippocampus, hypothalamus, and brainstem. Unlike most potassium channels it is constitutively active, setting baseline neuronal excitability rather than responding to action potentials. It is opened by multiple physiological stimuli including mechanical stretch, heat, acidosis, volatile anesthetics, and lipids. Its relevance to depression was established when studies showed that TREK-1 knockout mice — mice genetically lacking this channel — were resistant to depression-like behaviors in multiple validated animal models. Conversely TREK-1 overexpression promotes depression-like states. Human genetic association studies have linked TREK-1 variants to depressive disorders in over 750 patients. SSRIs were found to partially reduce TREK-1 activity, suggesting TREK-1 inhibition may be part of their mechanism — but with far less selectivity and much slower onset than direct TREK-1 antagonists.
PE-22-28 functions as a potent and selective antagonist of TREK-1. The published peer-reviewed study in PMC from the French group documented an IC50 of 0.12 nM for PE-22-28 against human TREK-1 expressed in HEK cells — compared to 40 to 60 nM for the parent spadin — representing a 300 to 500-fold improvement in potency. By blocking TREK-1, PE-22-28 increases neuronal excitability in mood-relevant circuits, promotes hippocampal neurogenesis — the growth of new neurons in the hippocampus, a brain region consistently found to be atrophied in depression — and normalizes serotonergic neurotransmission in a manner that appears mechanistically faster than SSRIs. Critically the effect appears to involve serotonin — depletion of serotonin reversed the depression-resistant phenotype in TREK-1 knockout mice — suggesting TREK-1 inhibition acts upstream of serotonin signaling rather than bypassing it entirely.
The primary published data comes from the French research group's work in rodent models. PE-22-28 and its analogs demonstrated antidepressant effects in validated mouse depression models including the forced swim test and chronic mild stress paradigm. Critically the onset of antidepressant effect appeared within days rather than weeks — contrasting with the 2 to 4 week delay typical of SSRIs. The modified PE-22-28 analogs with glycine-alanine substitutions showed in vivo half-life effects lasting approximately 14 to 23 hours depending on dose — substantially longer than spadin's 7 hours. Unlike SSRIs, PE-22-28 appeared devoid of the typical SSRI-associated side effects in animal models including effects on libido, sleep architecture, or appetite. Post-stroke depression models showed particularly promising results — PE-22-28 reduced depression-like behaviors and supported neurogenesis in animals that had experienced cerebral ischemia.
In research and wellness contexts PE-22-28 is given by subcutaneous injection. Research protocols typically cite doses in the range of a few micrograms per kilogram in animal studies. Human-translated dosing estimates used in wellness contexts range from 100 to 500 mcg per injection. Frequency varies by protocol — some suggest daily administration given the 14 to 23 hour effect duration, others suggest less frequent dosing. It is supplied as lyophilized powder reconstituted with bacteriostatic water. Intranasal administration has also been explored given the blood-brain barrier access advantage.
PE-22-28's side effect profile in the available animal research is notably clean compared to SSRIs — a key feature emphasized by its developers. No significant effects on appetite, sleep architecture, libido, memory, or motor function were observed in animal models at antidepressant doses. No serious adverse effects have been documented in preclinical studies. Given the complete absence of human clinical trial data, the human side effect profile is entirely unknown. Theoretical concerns from TREK-1 inhibition include potential effects on pain perception, cardiovascular function, and neuronal hyperexcitability at higher doses given TREK-1's roles in neuroprotection against epilepsy and ischemia — TREK-1 opening has been shown to be protective in epilepsy and stroke models, meaning sustained antagonism at inappropriate doses could theoretically increase neuronal vulnerability in these contexts.
People with active epilepsy or a history of seizures should not use it given TREK-1's neuroprotective role against epileptic activity — inhibiting this channel could theoretically lower seizure threshold. People with a history of stroke or significant cerebrovascular disease should exercise caution for similar reasons despite the post-stroke depression research suggesting benefit in animal models. Anyone with severe depression or suicidal ideation should seek established, evidence-based clinical treatment rather than experimental research compounds. Pregnant or breastfeeding women should not use it. It is not FDA-approved for any indication and should only be considered within formally supervised research contexts.